Physique - Energie de surface
 1. Origine physique et définitions
1. Origine physique et définitions
Introduction et connaissances de base
Une surface caractérise la discontinuité d’un volume de matière condensée. Géométriquement, elle représente la frontière, l’interface entre deux phases (solide-solide, solide-liquide, liquide-liquide, liquide-gaz ou solide-gaz). Cette discontinuité (zone de transition) lui confère des propriétés tout à fait particulières et différentes de celle des atomes du volume.
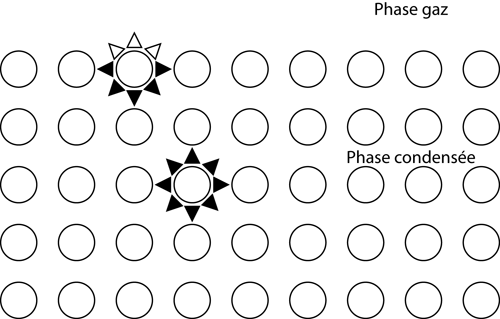
Si l’on considère les attractions positives entre les atomes (ou molécules) responsables de la cohésion d’une phase condensée, on peut remarquer (figure 1) la coordinence plus faible des atomes (ou molécules) de surface en comparaison à ceux du volume.
Cette coordinence plus faible à la surface que dans le volume indique que la création d’une surface est endothermique, qu’elle demande un apport énergétique. En considérant l’énergie interne ![]() par constituant du système et la dimension caractéristique
par constituant du système et la dimension caractéristique ![]() (atome ou molécule), une approche simplifiée permet d’écrire
(atome ou molécule), une approche simplifiée permet d’écrire
 où
où ![]() est l’énergie de surface par unité d’aire. C’est de cette approche que provient la définition la plus commune de l’énergie de surface :
est l’énergie de surface par unité d’aire. C’est de cette approche que provient la définition la plus commune de l’énergie de surface :
L’énergie de surface équivaut au travail nécessaire pour incrémenter de dA une surface d’aire A à température, volume et nombre de constituants constants.
Ce qui revient à écrire :
![]()
D’un point de vue thermodynamique, une surface doit donc être traitée comme une
grandeur en excès dans l’expression de l’énergie d’un corps [1]. Ainsi, en considérant une surface d’épaisseur nulle, on ajoute cette grandeur aux expressions de la variation d’énergie interne et de la variation d’énergie libre d’un système donné :
![]()
![]()
Avec :![]() , l’énergie interne (J)
, l’énergie interne (J)![]() , l’énergie libre de Helmholtz (J)
, l’énergie libre de Helmholtz (J)![]() , la température (K)
, la température (K)![]() , l’entropie (J·K−1)
, l’entropie (J·K−1)![]() , la pression (Pa)
, la pression (Pa)![]() , le volume (m3)
, le volume (m3)![]() , l’énergie de surface (J·m−2)
, l’énergie de surface (J·m−2)![]() , l’aire (m2)
, l’aire (m2)![]() , le potentiel chimique de l’espèce i (J·mol−1)
, le potentiel chimique de l’espèce i (J·mol−1)![]() , le nombre total de moles de l’espèce i (mol)
, le nombre total de moles de l’espèce i (mol)
L’énergie de surface peut alors être exprimée à partir des variations partielles d’énergie
interne à entropie, volume et nombre de moles constants ou d’énergie libre à température,
volume et nombre de moles constants :
_{S,V,n}=\left(\frac{\partial F}{\partial A}\right)_{T,V,n}")
Cet excédent d’énergie à la surface a pour conséquence de nombreux phénomènes. Thermodynamiquement, une surface est instable et tend naturellement à diminuer le produit ![]() . Une possibilité est la réduction du terme
. Une possibilité est la réduction du terme ![]() en modifiant la géométrie de la surface ce qui explique la tendance naturelle à la forme sphérique (le plus faible rapport aire sur volume) et les phénomènes de reconstruction à la surface. Pour les volumes comportant des phases à plusieurs constituants, les composés à la plus faible énergie de surface seront ségrégés vers celle-ci. Enfin, la présence d’une phase gazeuse au contact d’une surface engendre des phénomènes d’adsorption qui permettent également de baisser le produit
en modifiant la géométrie de la surface ce qui explique la tendance naturelle à la forme sphérique (le plus faible rapport aire sur volume) et les phénomènes de reconstruction à la surface. Pour les volumes comportant des phases à plusieurs constituants, les composés à la plus faible énergie de surface seront ségrégés vers celle-ci. Enfin, la présence d’une phase gazeuse au contact d’une surface engendre des phénomènes d’adsorption qui permettent également de baisser le produit ![]() . Ainsi, le travail de création d’une surface est irréversible. En pratique, nous avons donc affaire à ces surfaces « stabilisées », ayant minimisé leur énergie. Cette notion est importante et il est nécessaire de la garder à l’esprit dans la mesure où l’approche thermodynamique concerne des surfaces fictives (propres et vierges) et
. Ainsi, le travail de création d’une surface est irréversible. En pratique, nous avons donc affaire à ces surfaces « stabilisées », ayant minimisé leur énergie. Cette notion est importante et il est nécessaire de la garder à l’esprit dans la mesure où l’approche thermodynamique concerne des surfaces fictives (propres et vierges) et
que l’étude de surfaces fonctionnalisées concerne des surfaces réelles.
[1] R. J. GOOD. Physical signifiance of parameters ![]() ,
, ![]() and
and ![]() , that govern spreading on adsorbed films. In Society of Chemical Industry Monograph, volume 25, pages 328–350. Marcel Dekker, Inc., 1967.
, that govern spreading on adsorbed films. In Society of Chemical Industry Monograph, volume 25, pages 328–350. Marcel Dekker, Inc., 1967.
Avertissement
Le contenu de cet article est une reproduction numérique en ligne de la thèse de Mathias Borella soutenue en octobre 2006. Seules les informations délivrées dans l'édition papier du manuscrit font foi, sont valides et vérifiées. En cas de doute, merci de vous y référer.
Cet article a été publié en ligne pour la première fois en Février 2009. Il a été passé en revue pour la dernière fois le 5 février 2009.